جمعه, ۲۴ اسفند, ۱۴۰۳ / 14 March, 2025
مجله ویستا
کاربرد میکرو آرایه در بیان ژن
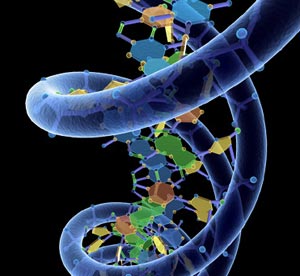
پروتئینها مولکولهای بزرگی هستند، که اساس هر ارگانیزمی را تشکیل میدهند. همه سلولها در یک ارگانیزم ژنهای مشابهی دارند، اما این ژن ها در زمانهای مختلف و در شرایط مختلف، بیان متفاوتی دارند.
در سال های اخیر تکنولوژی میکروآرایه امکان مانیتورینگ بیان هزاران ژن به صورت همزمان را فراهم کرده و ژنومیک و پروتئومیک اساسا شیوه علمی مطالعه پایه مولکولی رفتارهای بافت و سلول در شرایط فیزیولوژیک و پاتولوژیک را تغییر داده و یک شیوه فراگیر در اختیار جامعه تحقیقاتی قرار دادهاند . به طور خاص تکنیک میکروآرایه برای پروفایل همزمان بیان هزاران ژن در بازه وسیعی از تحلیلهای ژنومیک، نظیر شناسائی ژن ها، اکتشاف داروها و تشخیصهای کلنیکی به صورت موفقیت آمیزی مورد استفاده قرار گرفتهاند.
این تکنیک امکان فهم فرایندهای سلولی و شبکههای نسخهبرداری را به صورت genome-wide system-level فراهم میکند.[۱] الگوی بیان ژن یک سلول یا یک بافت، ساختار و عملکرد آن را تعیین میکند. بیان ژن فرایندی دینامیک است، که میتواند به صورت گذرا یا دائمی تغییر کند، بنابراین میتواند تغییرات لحظهای و ماندگاری در حالت بیولوژیک سلولها، بافتها، اعضا و ارگانیزم ها را منعکس کند.[۲] دادههای سطوح بیان ژنها اطلاعات ارزشمندی در مورد شبکههای بیولوژیک، حالات سلولی و فهم چگونگی ژنها در بردارد.
یک هدف از تحلیل داده بیان ژن تعیین چگونگی تأثیر بیان هر ژن منفرد روی بیان ژن های دیگر در همان شبکه ژنتیکی است. هدف دیگر مشخص کردن این نکته است، که چگونه ژنها در سلولهای سالم و بیمار بیان میشوند.کاربرد عملی پروفایل بیان ژن میکروآرایه مدیریت و کنترل سرطان و بیماریهای عفونی است.[۳] هدف اصلی این مطالعات، تعیین و شناسائی فرایند پاتولوژیک مرتبط با نوع بیماری و مرحله آن و نیز پیشبینی بیماری و پیش بینی پاسخ به درمان خاصی، است.
ممکن است برخی از مسائل مشکل در زمینه تشخیص، با استفاده از تحلیل دادههای بیان ژن حل شوند. به عنوان مثال تومورهای Gastrointestinal stromal ) GISTs)طیف وسیعی از ابتدای تشکیل تومور تا تومورهای بدخیم را در برمیگیرند که تشخیص آن بسیار مشکل است. در حال حاضر استاندارد طلائی تشخیص GIST، KIT immunohistochemistry است، اما تعدادی از تومورها در KIT تشخیص داده نمیشوند. مطالعات جدید نشان داده است که protein kinase C theta در GIST بیان بالایی دارد، در حالی که در تومورهای دیگر mesenchymal نیست. بنابراین احتمالا تشخیص این ژن میتواند یک روش مکمل در تشخیص این بیماری باشد.[۲]
در روش های کلاسبندی سرطان بر اساس علائم مورفولوژیکی ممکن است انواع مختلف سرطان، نشانههای مورفولوژیک مشابهی داشته باشند، لذا نتوان بر این اساس کلاس بندی درستی انجام داد. اما از آنجا که هر بیماری، از جمله یک تومور اساسا عملکرد بد ژنها است، استفاده از دادههای بیان ژن میتواند شیوههای تشخیصی مستقیمی را فراهم کند.
آزمایشهایی که بر اساس تکنولوژی میکروآرایه انجام میشوند حجم بسیار زیادی از دادهها را فراهم میکنند که در مطالعات بیولوژیک بی نظیر بوده است. این دادهها میتوانند برای جامعه تحقیقات کلی معدنی از اطلاعات باشند[۱]. برای بررسی حجم وسیع اطلاعات تولید شده و استخراج اطلاعات مفید از آنها و استفاده از آنها در سالهای اخیر تلاشهای فراوانی صورت گرفته است. تفسیر دادههای بیان ژن به دست آمده از میکروآرایه که پیچیده و نویزی هستند بسیار مهم است، چرا که دادههای میکروآرایه DNA بدون وجود ابزاری برای تحلیل آنها بدون استفاده خواهند بود.
المان اصلی در تکنولوژی میکروآرایه، آرایه ای از پروبهای مولکولی است که مکمل توالیهای خاصی از mRNA است و روی یک فاز جامد، به عنوان مثال یک اسلاید شیشهای، ثابت شده است. پروبها میتوانند با استفاده از تکنیک فوتولیتوگرافیک ، مستقیما روی فاز جامد سنتز شوند(Affymetrix) یا اینکه روی فاز جامد پرینت شوند.(محصولات PCR یا الیگونوکلئوتید).
این سیستمها تا ۱۰۰۰۰۰ probes/cm۲ را تامین میکنند، که امکان بررسی وسیع بیانهای ژن رافراهم میکند.mRNA نمونهها استخراج برچسب گذاری و با میکروآرایه ترکیب میشوند و با استفاده از تکنیکهای مختلف شناسائی میشوند، که بیشتر این تکنیکها بر پایه خواندن نشانگرهای فلورسانس عمل میکنند. پرکاربردترین محصولات میکروآرایه درحال حاضر چیپهای Affymetrix هستند .فرآیند کلی که در آن با استفاده از میکروآرایه ها، دادههای بیان ژن به دست آورده میشوند در شکل نشان داده شده است.
آنالیز پایه دادههای بیان ژن شامل یک مرحله پیش پردازش و آماده سازی مجموعه داده برای مراحل آنالیز سطوح بالاتر مانند خوشه بندی یا کلاس بندی است. پیش پردازش دادههای خام میتواند اثرات عمیقی روی مراحل بعدی آنالیزداشته باشد.
مراحل پیشپردازشی که روی دادههای میکروآرایه انجام میشوند، عبارت است از:
▪ مقیاس گذاری:
معمولا دادههای خام بیان ژن از چندین نمونه ("چیپ") به منظور جبران اثر تغییرات کلی ناشی از اختلاف شدتهای چیپ و تفاوت از یک میکروآرایه به میکروآرایه دیگر درجهبندی میشوند.
آستانه گذاری، فیلتر کردن و نرمال کردن: ازآنجا که دادههای بیان بسیار بالا یا بسیار پائین کمتر مورد اعتماد و تکرارپذیر هستند، لذا از آستانه گذاری داده استفاده میشود.
در ضمن بسیاری از الگوریتمهای خوشه بندی و کلاس بندی با تعداد کم دادهها بهتر کار میکنند و به دادههای نویزی حساس هستند، لذا ژنهائی که بیان آنها بین چند نمونه تغییر نمیکند یا این که بیان کمی در بین چند نمونه دارند، از مجموعه داده فیلتر میشوند. یکی از سادهترین روش هایی که برای این منظور استفاده میشود این است که بین نمونهها ماکزیمم و مینیمم بیان یک ژن خاص به دست میآید و اگر max / min و max - min ازمقادیر آستانهای کمتر بودند، ژنهای متناظر آنها از مجموعه داده حذف میشوند.
به طور کلی در ژنها اطلاعات مفیدی برای کلاس بندی سرطانها وجود دارد، اما اطلاعات غیر مفید نیز در آنها وجود دارد. لذا ویژگیهای مفید حتماً باید استخراج شوند. یک ژن در صورتی در یک بیماری خاص مناسب است که قادر باشد به درستی شرایط پاتولوژیک در بیماران جدید را بر پایه فقط سطح بیان ژن آن تشخیص دهد. به عبارت دیگر، یک ژن برای یک بیماری حاوی اطلاعات مفید است اگر سطوح بیان آن برای تعلیم یک طبقه بندی کننده مفید باشد و قدرت تعمیم را هم داشته باشد، یعنی بتواند به درستی وضعیت بیماران جدید را پیشگوئی کند[۷].
انتخاب ژنهای مناسب برای تشخیص و پیشگوئی سرطان به چند دلیل محاسباتی و بیولوژیک ارزشمند است. یافتن ژنهائی که سطوح بیان آنها با یک بیماری خاص همبستگی دارد برای انتخاب مناسب ترین درمان و پیش بینی بازگشت مجدد بیماری مهم است. در ضمن این امکان فراهم میشود که میکروآرایههای تک کاره و اقتصادیتر طراحی شوند که مناسب بیماری خاصی است و سطوح بیان فقط چند ده ژن را ثبت میکنند.
به علاوه انتخاب یک زیر مجموعه از ژنها امکان overfitting که با افزایش تعداد ویژگیها( سطوح بیان ژن) نسبت به تعداد نمونهها احتمال آن زیاد میشود را کاهش میدهد. [۷]
▪ نرمالیزاسیون:
بعد از فیلتر کردن عمل نرمالیزاسیون انجام میشود تا میانگین صفر و واریانس یک روی همه نمونههابه دست آید. این استراتژی در صورتی مفید است که آنچه مورد توجه کاربر است، تفاوت نسبی و نه مطلق بین ژنها باشد.
آنالیز سطوح بالاتر دادههای بیان ژن، یادگیری با و بدون سرپرست به روی دو شیوه متمرکز شده است:
ـ یادگیری بدون سرپرستی یا خوشه بندی:
درتکنیکهای یادگیری بدون سرپرستی، ساختار یک مجموعه داده بدون هیچ فرض یا دانش قبلی شناسایی میشود. یکی از سادهترین شیوهها استفاده از الگوریتمهای خوشه بندی است. افراد مورد نظر(ژنها یا نمونه ها) بر اساس میزان "نزدیکی" به هم به گروههائی طبقه بندی میشوند که این کار با استفاده از یک تابع "شباهت" یا همبستگی و یا فاصله بین نمونههاانجام میشود. وقتی در مورد داده ها، دانش قبلی وجود دارد میتوان نتایج خوشه بندی را تفسیر کرد. اما در صورت عدم وجود چنین دانشی تفسیر بیولوژیکی نتایج خوشه بندی یک چالش باقی میماند.
ـ یادگیری با سرپرستی از دانش مربوط به برچسب های کلاسها استفاده میکند تا ساختار مورد نظر را بسازد. از مجموعه داده ها آموزشی برای انتخاب ویژگی هائی که بتوانند بهترین طبقه بندی را بسازند، استفاده میشود. سپس این ویژگیهابه یک مجموعه داده تست مستقل اعمال میشوند تا توانائی ویژگیهای انتخاب شده در طبقه بندی ارزیابی یادگیری با سرپرست وابسته به این است که برچسبگذاری نمونهها به درستی انجام شود. فرایند کلی در کلاسیفایرهای دادههای بیان ژن میکروآرایه در دیاگرامهای زیر آورده شده است: [۶]
به منظور ارزیابی نتایج به دست آمده از تحلیل دادههای میکروآرایه، مقادیر زیادی از دادهها باید نگهداری شده و به روشی مناسب در دسترس قرار بگیرند. برای کم کردن مسائل نگهداری و sharing داده، در حال تدوین استانداردهائی هستند. در حال حاضر جامعه داده بیان ژن میکروآرایه (MGED) با ارائه استانداردهائی نظیر MIAME و MAGE MAGEOM و MAGE-ML، تلاشهای مستمری برای استانداردسازی دارد[۱]
فهرست منابع:
[۱] Francesco Beltrame, Adam Papadimitropoulos, Ivan Porro_, Silvia Scaglione, Andrea Schenone,Livia Torterolo, Federica Viti,” GEMMA — A Grid environment for microarray management and analysis in bone marrow stem cells experiments ”, Received ۳۱ January ۲۰۰۶; received in revised form ۲۱ April ۲۰۰۶; accepted ۲ July ۲۰۰۶ , Future Generation Computer Systems
[۲] A.K. Sandvik , B.K. Alsberg, K.G. Nørsett, F. Yadetie, H.L. Waldum, A. Lægreid,” Gene expression analysis and clinical diagnosis”, Clinica Chimica Acta ۳۶۳ (۲۰۰۶) ۱۵۷ – ۱۶۴
[۳] Shinn-Ying Ho, Chih-Hung Hsieh , Hung-Ming Chen, Hui-Ling Huang , “Interpretable gene expression classifier with an accurate and compact fuzzy rule base for microarray data analysis ”, BioSystems ۸۵ (۲۰۰۶) ۱۶۵–۱۷۶
[۴] Jin-Hyuk Hong, Sung-Bae Cho, “The classification of cancer based on DNA microarray data that uses diverse ensemble genetic programming ”, Artificial Intelligence in Medicine (۲۰۰۶) ۳۶, ۴۳—۵۸
[۵] Leonardo Angelini, Daniele Marinazzo, Mario Pellicoro, Sebastiano Stramaglia,” Kernel method for clustering based on optimal target vector”, Available online ۴ May ۲۰۰۶, Physics Letters A ۳۵۷ (۲۰۰۶) ۴۱۳–۴۱۶
[۶] Jin-Hyuk Hong, Sung-Bae Cho,” Efficient huge-scale feature selection with speciated genetic algorithm ” , Pattern Recognition Letters ۲۷ (۲۰۰۶) ۱۴۳–۱۵۰
[۷] Rosalia Maglietta , Annarita D’Addabbo , Ada Piepoli ,Francesco Perri , Sabino Liuni , Graziano Pesole , Nicola Ancona,” Selection of relevant genes in cancer diagnosis based on their prediction accuracy”, Artificial Intelligence in Medicine (۲۰۰۶) xxx, xxx—x
[۱] Francesco Beltrame, Adam Papadimitropoulos, Ivan Porro_, Silvia Scaglione, Andrea Schenone,Livia Torterolo, Federica Viti,” GEMMA — A Grid environment for microarray management and analysis in bone marrow stem cells experiments ”, Received ۳۱ January ۲۰۰۶; received in revised form ۲۱ April ۲۰۰۶; accepted ۲ July ۲۰۰۶ , Future Generation Computer Systems
[۲] A.K. Sandvik , B.K. Alsberg, K.G. Nørsett, F. Yadetie, H.L. Waldum, A. Lægreid,” Gene expression analysis and clinical diagnosis”, Clinica Chimica Acta ۳۶۳ (۲۰۰۶) ۱۵۷ – ۱۶۴
[۳] Shinn-Ying Ho, Chih-Hung Hsieh , Hung-Ming Chen, Hui-Ling Huang , “Interpretable gene expression classifier with an accurate and compact fuzzy rule base for microarray data analysis ”, BioSystems ۸۵ (۲۰۰۶) ۱۶۵–۱۷۶
[۴] Jin-Hyuk Hong, Sung-Bae Cho, “The classification of cancer based on DNA microarray data that uses diverse ensemble genetic programming ”, Artificial Intelligence in Medicine (۲۰۰۶) ۳۶, ۴۳—۵۸
[۵] Leonardo Angelini, Daniele Marinazzo, Mario Pellicoro, Sebastiano Stramaglia,” Kernel method for clustering based on optimal target vector”, Available online ۴ May ۲۰۰۶, Physics Letters A ۳۵۷ (۲۰۰۶) ۴۱۳–۴۱۶
[۶] Jin-Hyuk Hong, Sung-Bae Cho,” Efficient huge-scale feature selection with speciated genetic algorithm ” , Pattern Recognition Letters ۲۷ (۲۰۰۶) ۱۴۳–۱۵۰
[۷] Rosalia Maglietta , Annarita D’Addabbo , Ada Piepoli ,Francesco Perri , Sabino Liuni , Graziano Pesole , Nicola Ancona,” Selection of relevant genes in cancer diagnosis based on their prediction accuracy”, Artificial Intelligence in Medicine (۲۰۰۶) xxx, xxx—x
منبع : مجله مهندسی پزشکی و تجهیزات آزمایشگاهی













































































ایران مسعود پزشکیان دولت چهاردهم پزشکیان مجلس شورای اسلامی محمدرضا عارف دولت مجلس کابینه دولت چهاردهم اسماعیل هنیه کابینه پزشکیان محمدجواد ظریف
پیاده روی اربعین تهران عراق پلیس تصادف هواشناسی شهرداری تهران سرقت بازنشستگان قتل آموزش و پرورش دستگیری
ایران خودرو خودرو وام قیمت طلا قیمت دلار قیمت خودرو بانک مرکزی برق بازار خودرو بورس بازار سرمایه قیمت سکه
میراث فرهنگی میدان آزادی سینما رهبر انقلاب بیتا فرهی وزارت فرهنگ و ارشاد اسلامی سینمای ایران تلویزیون کتاب تئاتر موسیقی
وزارت علوم تحقیقات و فناوری آزمون
رژیم صهیونیستی غزه روسیه حماس آمریکا فلسطین جنگ غزه اوکراین حزب الله لبنان دونالد ترامپ طوفان الاقصی ترکیه
پرسپولیس فوتبال ذوب آهن لیگ برتر استقلال لیگ برتر ایران المپیک المپیک 2024 پاریس رئال مادرید لیگ برتر فوتبال ایران مهدی تاج باشگاه پرسپولیس
هوش مصنوعی فناوری سامسونگ ایلان ماسک گوگل تلگرام گوشی ستار هاشمی مریخ روزنامه
فشار خون آلزایمر رژیم غذایی مغز دیابت چاقی افسردگی سلامت پوست